

Gene type of a patient with Kartagener syndrome and literature review
-
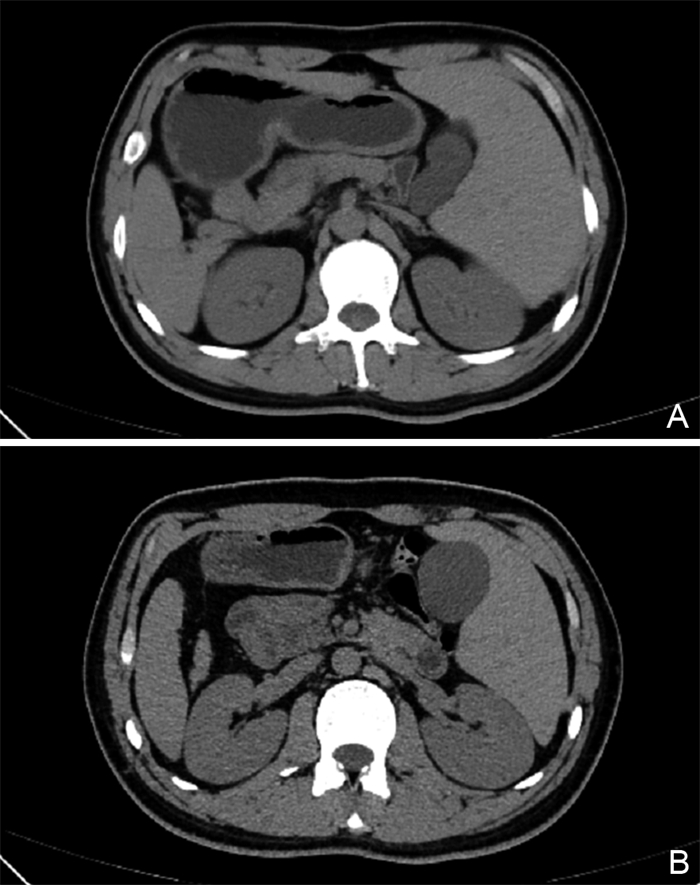
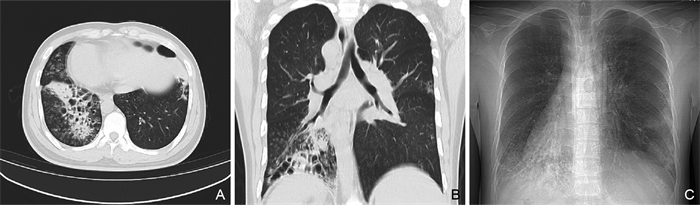
摘要: 本文回顾性分析1例Kartagener综合征(Kartagener Syndrome,KS)病例的临床资料,检索数据库对近10年报道的KS病例进行分析归纳。患者男性,23岁,自幼反复发作鼻窦炎、肺部感染。完善辅助检查提示全组副鼻窦炎、支气管扩张并感染、右位心、腹腔全脏器反位。通过基因检测技术,在原发性纤毛运动障碍3型伴或不伴内脏异位相关的DNAH5基因上检出2个与受检者表型相符的致病变异;在常染色体隐性遗传的原发性纤毛运动障碍6型相关的NME8基因上检出2个与受检者表型相符的罕见变异,确诊为KS。全科医生应重视KS等遗传病和罕见病,通过文献检索扩大诊疗思路,辅以基因检测技术协助临床诊断,做到早期诊断,早期管理,贯彻以患者为中心的全科理念。
-
关键词:
- Kartagener综合征 /
- 支气管扩张 /
- 右位心 /
- 内脏异位 /
- 全科医学科
Abstract: The clinical data of one Kartagener syndrome (KS) case were retrospectively analyzed, and the reported KS cases in the past 10 years were analyzed and summarized by searching the database. The patient was a 23-year-old male with recurrent sinusitis and pulmonary infection since childhood. Complete auxiliary examination showed the whole group of paranasal sinusitis, bronchiectasis with infection, dextrocardia, and peritoneal situs inversus totalis. Two pathogenic variants were detected in DNAH5 gene of primary ciliary dyskinesia type 3 with or without splanchnic ectopic. Two rare variants in the NME8 gene associated with autosomal recessive inheritance primary ciliary dyskinesia type 6 were identified, which was diagnosed as KS. General practitioners should pay attention to genetic diseases and rare diseases such as KS, expand diagnosis and treatment ideas through literature retrieval, assist clinical diagnosis with genetic testing technology, achieve early diagnosis and early management, and implement the patient-centered philosophy of general practice.-
Key words:
- Kartagener syndrome /
- Bronchiectasis /
- Dextrocardia /
- Splanchnectopia /
- Department of general practice
-
表 1 Kartagener综合征患者及父母基因检测结果
Table 1. Genetic testing results of patients and their parents with Kartagener syndr
基因 转录本 染色体位置 核苷酸改变 氨基酸改变 1000g/ESP/ExAC S/Mt/Ms/P 纯合/杂合 变异来源 DNAH5 NM_001369 chr5:13841077 c.5647C>T p.Arg1883Ter N/A|N/A|4.121e-05 .|A|.|. 杂合 父亲 DNAH5 NM_001369 chr5:13842004 c.5281C>T p.Arg1761Ter N/A|N/A|4.136e-05 .|A|.|. 杂合 母亲 NME8 NM_016616 chr7:37907475 c.793G>T p.Gly265Ter 0.0006|N/A|0.0001 .|N|.|. 杂合 父亲 NME8 NM_016616 chr7:37907332 c.650T>C p.Met217Thr 0.0006|N/A|0.0001 D|D|D|D 杂合 父亲 注:1000g为1000 Genomes,千人基因组数据库;ESP为NHLBI Exome Sequencing Project,NHLBI外显子组测序项目;ExAC为Exome Aggregation Consortium,外显子集成联合。A(Disease causing automatic)表示有害,D(Disease causing)表示可能有害,N(Polymorphism)表示可能无害,P(Polymorphism automatic)表示无害,其中A和P表示该变异在已知数据库中有记录,证据明显。黑点表示空。  下载: 导出CSV
下载: 导出CSV
表 2 ACMG指南解读变异临床意义
Table 2. Clinical significance of variants in ACMG guidelines
项目 变异类型 PVS1 p.Arg1883Ter为无义变异,会导致DNAH5基因功能丧失,功能丧失为DNAH5基因的致病。 PM2 1 000g、ESP数据库、ExAC数据库中正常对照人群中未发现的变异(或隐性遗传病中极低频位点)。 PP5 有可靠信誉来源的报告认为该变异为致病的,但证据尚不足以支持进行实验室独立评估。
下载: 导出CSV
-
[1] 张静, 白银, 尤少华, 等. Kartagener综合征合并分泌性中耳炎患者的基因诊断[J]. 中华耳科学杂志, 2014, 12(1): 41-44. https://www.cnki.com.cn/Article/CJFDTOTAL-ZHER201401012.htmZHANG J, BAI Y, YOU S H, et al. Genetic diagnosis of Kartagener syndrome patients with secretory otitis media[J]. Chinese Journal of Otology, 2014, 12(1): 41-44. https://www.cnki.com.cn/Article/CJFDTOTAL-ZHER201401012.htm [2] 王文淼, 马宁, 宁君. 儿童完全性Kartagener综合征1例并文献复习[J]. 临床肺科杂志, 2019, 24(9): 1738-1741. doi: 10.3969/j.issn.1009-6663.2019.09.045WANG W M, MA N, NING J. A case of complete Kartagener syndrome in children and literature review[J]. Journal of Clinical Pulmonary Medicine, 2019, 24(9): 1738-1741. doi: 10.3969/j.issn.1009-6663.2019.09.045 [3] 王珂. 原发性纤毛运动障碍纤毛超微结构与基因型分析[D]. 济南: 山东大学临床医学院, 2017.WANG K. Analysis of ultrastructure and genotype of cilia in primary ciliary dyskinesia[D]. Jinan: Shandong University School of Medicine, 2017. [4] 谢鼎华. 耳鼻咽喉临床遗传学[M]. 长沙: 湖南科技出版社, 2002: 228.XIE D H. Clinical genetics of otorhinolaryngology[M]. Changsha: Hunan Science and Technology Press, 2002: 228. [5] 李华, 施蓉萍. Kartagener综合征1例并文献复习[J]. 中华肺部疾病杂志(电子版), 2012, 5(6): 575-576. https://www.cnki.com.cn/Article/CJFDTOTAL-ZFBD201206034.htmLI H, SHI R P. A case of Kartagener syndrome and literature review[J]. Chinese Journal of Pulmonary Diseases (Electronic Edition), 2012, 5(6): 575-576. https://www.cnki.com.cn/Article/CJFDTOTAL-ZFBD201206034.htm [6] FAILLY M, BARTOLONI L, LETOURNEAU A, et al. Mutations in DNAH5 account for only 15% of a non-preselected cohort of patients with primary ciliary dyskinesia[J]. J Med Genet, 2009, 46(4): 281-286. doi: 10.1136/jmg.2008.061176 [7] ZARIWALA M A, GEE H Y, KURKOWIAK M, et al. ZMYND10 is mutated in primary ciliary dyskinesia and interacts with LRRC6[J]. Am J Hum Genet, 2013, 93(2): 336-345. doi: 10.1016/j.ajhg.2013.06.007 [8] HORNEF N, OLBRICH H, HORVATH J, et al. DNAH5 mutations are a common cause of primary ciliary dyskinesia with outer dynein arm defects[J]. Am J Respir Crit Care Med, 2006, 174(2): 120-126. doi: 10.1164/rccm.200601-084OC [9] OLBRICH H, HAFFNER K, KISPERT A, et al. Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left-right asymmetry[J]. Nat Genet, 2002, 30(2): 143-144. doi: 10.1038/ng817 [10] DANIELS M L A, NOONE P G. Genetics, diagnosis, and future treatment strategies for primary ciliary dyskinesia[J]. Exper Opin Orphan Drugs, 2015, 3(1): 3144. [11] JANE S L, ANDREA B, HANNAH M M, et al. Diagnosis and management of primary ciliary dyskinesia[J]. Arch Dis Child, 2014, 99(9): 850-856. doi: 10.1136/archdischild-2013-304831 [12] GEREMEK M, WITT M. Primary ciliary dyskinesia: Genes, candidate genes and chromosomalrgions[J]. J Applied Genetics, 2004, 45(3): 347-361. [13] MORILLAS H N, ZARINWALA M, KNOWLES M R. Genetic cause of bronchiectasis: Primary ciliary dyskinesia[J]. Respiration, 2007, 74(3): 252-263. doi: 10.1159/000101783 [14] LUCAS J S, BURGESS A, MITCHISON H M, et al. Diagnosis and management of primary ciliary dyskinesia[J]. Arch Dis Child, 2014, 99(9): 850-856. [15] FAIQA I, RABAB A, KHUSHNOODA R, et al. Variation in DNAH1 may contribute to primary ciliary dyskinesia[J]. Bmc Medical Genetics, 2015, 16: 14. -

 点击查看大图
点击查看大图
图(3) / 表(2)
计量
- 文章访问数: 206
- HTML全文浏览量: 125
- PDF下载量: 23
- 被引次数: 0
